Por Débora Carvalho Meldau
A doença de Huntington, também chamada de “coréia de Huntington”, é uma doença degenerativa progressiva do sistema nervoso com padrão de herança autossômico dominante de penetrância completa. Deste modo, a progênie de um indivíduo afetado, possui 50% de probabilidade de apresentar a doença.
Foi descrita pelo médico norte-americano George Huntington, em 1872, vindo dele o nome da afecção. Nas últimas décadas essa doença tem sido bastante estudada, sendo que o gene causador da doença (localizado próximo a extremidade telomérica do braço curto do cromossomo 4) foi descoberto em 1983, por meio da análise de ligações.
Esta doença está presente em todo o mundo; contudo, em certas regiões, como, por exemplo, a região do Maracaibo, na Venezuela, apresenta uma elevada incidência. Nos Estados Unidos, estima-se que sua prevalência encontre-se entre 5 a 10 casos por 100.000 habitantes. A idade média do surgimento da doença é de 40 anos, apesar de já ter sido observada aos 2 anos de idade e aos 80 anos de idade.
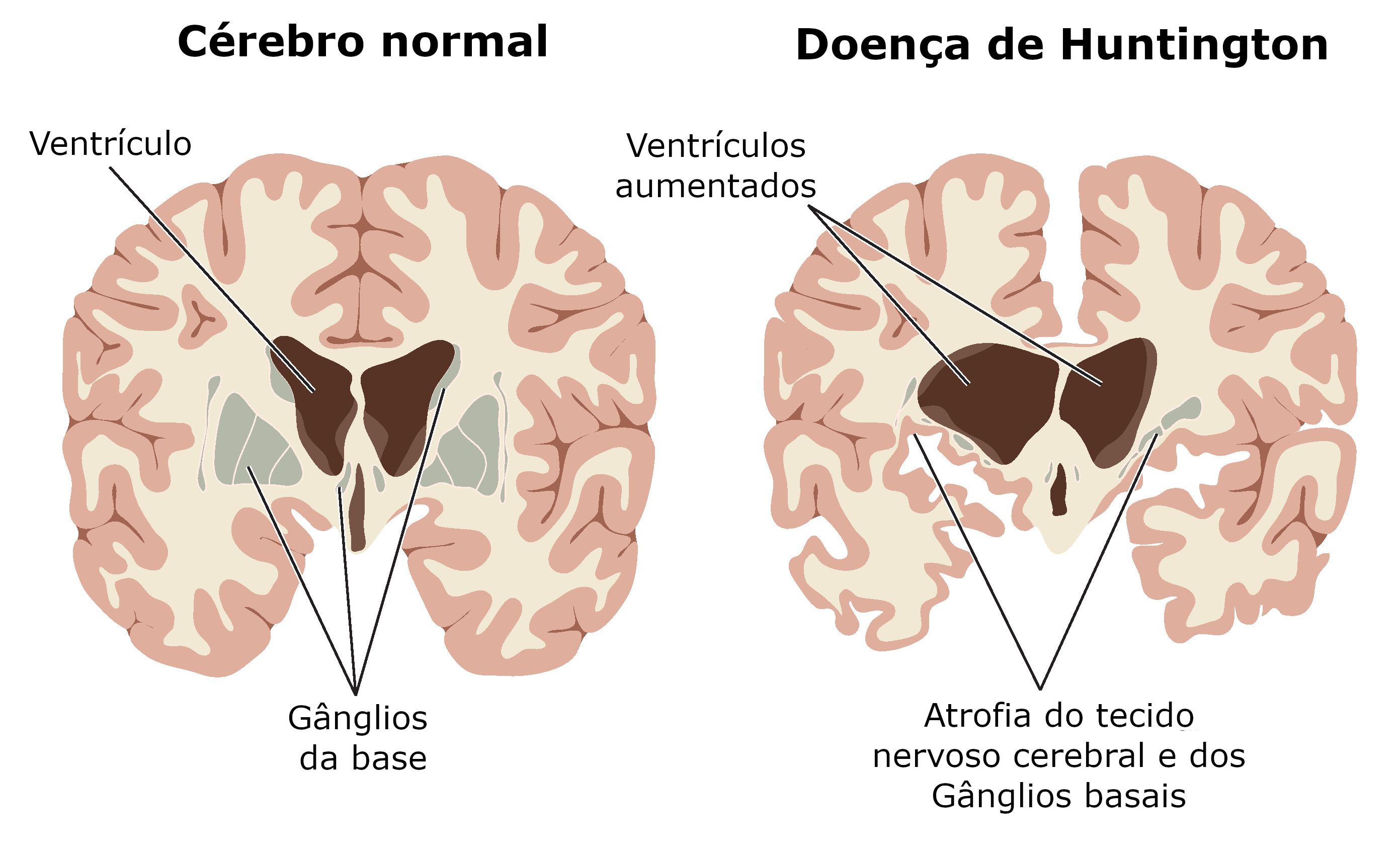
Ilustração mostra comparativo entre um cérebro normal e o cérebro de um portador da Doença de Huntington. Créditos: Blamb / Shutterstock.com (adaptado)
Os sinais clínicos mais comuns da doença são os movimentos involuntários (discinesia) dos braços, pernas e rosto, acometendo também aspectos da personalidade e habilidades mentais. Além desses sintomas, o paciente pode apresentar depressão, disartria, fala indistinta, dificuldade de mastigar e deglutir alimentos e perda da visão periférica.
O diagnóstico baseia-se no quadro clínico característico e histórico familiar. O diagnóstico definitivo é feito apenas por meio de exame genético. Por meio de exames de imagem é possível notar alterações neuroanatômicas.
O diagnóstico diferencial é bem amplo, incluindo enfermidades como a coréia familiar benigna, neuroacantocitose, atrofia dentatorubropalidolusiana (DRPLA), doença de Machado-Joseph (SCA-3), doenças priônicas e uma doença reconhecida recentemente chamada Huntington-like 2.
Atualmente não há ainda um tratamento preventivo ou curativo para a doença de Huntington, sendo que este é apenas sintomático. O tratamento com certos fármacos, como bloqueadores dos receptores dopaminérgicos (fenotiazinas ou haloperidol), pode controlar os movimentos involuntários e alguns dos distúrbios comportamentais. No entanto, esses medicamentos podem induzir um quadro tardio de discinesia superposta ao distúrbio crônico, devendo ser utiliza apenas em situações absolutamente necessárias. Indica-se o tratamento dessa doença apenas em casos incapacitante, utilizando-se mínimas doses de algum desses medicamentos, em dias alternados. O uso de agentes antidepressivos e ansiolíticos pode ser benéfico para alguns pacientes.
Leia mais:
Texto originalmente publicado em https://www.infoescola.com/doencas/doenca-de-huntington/